We recommend CellFunTopic package to perform data
pre-processing for convenience. The CellFunTopic
package provides a convenient workflow for data pre-processing (quality
control, normalization, dimension reduction, clustering, differential
expression analysis, etc.) by integrating methods of Seurat
package.
devtools::install_github("compbioNJU/CellFunTopic")
library(CellFunTopic)Load in the data
CellFunTopic allows various types of input, including CellRanger
outputs, raw gene expression matrix, and popular R objects used for
single cell analysis such as SingleCellExperiment,
Seurat, CellDataSet. CellFunTopic will
transform different input data into a Seurat object.
Let us use a randomly generated tiny single-cell expression matrix of Arabidopsis thaliana as a toy example.
## [1] 7000 2000
SeuratObj <- readData(data = example_Ath,
type = 'expMatrix',
species = "Arabidopsis thaliana")You can also take CellRanger outputs as input, for example:
SeuratObj <- readData(data = "filtered_gene_bc_matrices/TAIR/",
type = '10X',
species = "Arabidopsis thaliana")Standard pre-processing workflow
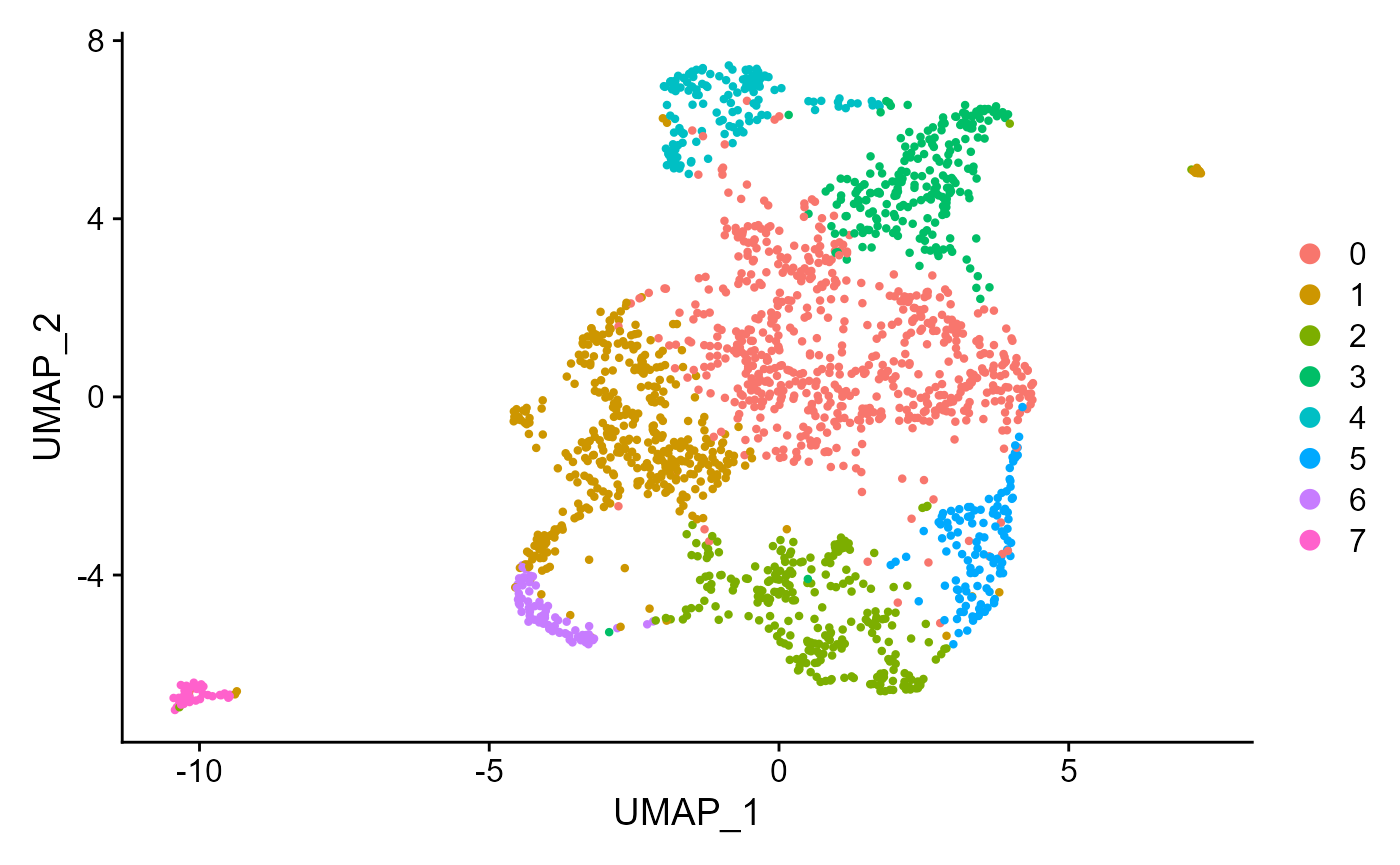
Then we can perform pre-processing conveniently. If users provide a pre-processed Seurat object, this step can be skipped.
## [1] 1 0 3 2 5 4 6 7
## Levels: 0 1 2 3 4 5 6 7
Seurat::DimPlot(SeuratObj, reduction = "umap")